There is widespread agreement that the United States Food and Drug Administration (FDA) needs reforming. The drug approval process in the United States is too slow, too expensive and too restrictive. The FDA delays the introduction of new drugs for up to 12 years and does not publish standards of safety or effectiveness that any drug can meet to ensure its approval. As a result:
- Thousands of patients die because lifesaving drugs and medical devices available elsewhere are not yet approved for use in the United States.
- Surgery, in some cases the only alternative to drug therapy, adds to the pain and suffering.
- Pharmaceutical companies are moving drug testing and development facilities out of the U.S. to reduce the costs imposed on them by the FDA approval process.
Contrary to its recent claims of improved efficiency, the FDA still delays drug development and approval, while rejecting more applications for approval of new drugs.
Congress is considering substantial reforms that include elements of the European drug and medical device approval system: self-certification by manufacturers, certification by public or private third parties and independent review committees. However, these reforms would leave FDA bureaucrats in control of drug approvals or increase political influence over the process.
The best way to make the process more efficient while preserving the safety and effectiveness of drugs is to adopt a market-based system. In such a system, the FDA would have little or no role, and private, independent third parties would certify all drugs and devices.
The model for a market-based drug certification system is Underwriters Laboratories, Inc. (UL), an independent, not-for-profit safety certification and standards-writing organization. For more than 100 years, UL has certified the safety of products on which millions of Americans rely. It shows how the marketplace can preserve third-party independence, since UL, consumers and manufacturers all have incentives to support high standards as well as fast, efficient certifications.
Manufacturers are not legally required to seek UL approval, but tens of thousands do. Product makers want to meet recognized standards in order to satisfy consumers and because they are legally liable for unsafe products. Competition between UL and other organizations ensures the integrity of the certification process. Even government agencies use UL standards:
- More than 40,000 local jurisdictions across the U.S. accept the UL mark and work with UL to develop electrical, building and safety codes.
- The federal Occupational Safety and Health Administration uses UL to independently test and certify products for hazardous locations, works with UL to revise and implement product standards and in some cases even uses UL's standards.
Private certification would allow people in different circumstances to balance the risks they choose to accept. Under such a system, a drug might be available without third-party certification, but doctors would be very reluctant to prescribe it. The only consumers who would use such drugs might be those willing to take great risks, such as terminally ill AIDS and cancer patients, but no one would be denied access to lifesaving drugs.
Private certification would be faster, less expensive and just as safe as the current FDA system.
[page]1The United States Food and Drug Administration (FDA) is the federal agency charged by law with protecting the public by approving drugs and medical devices before they are offered for sale in the U.S. Unfortunately, satisfying the FDA that a new drug is safe and effective delays the introduction of new drugs for up to 12 years and denies thousands of Americans access to potentially lifesaving therapies.
The FDA needs reform – manufacturers, consumer groups, academics, Congress and even the FDA itself realize this. Drug and medical device approval in the United States simply takes too long.
"Drug and medical device approval in the United States simply takes too long."
Some proposed reforms would apply the European system of approving drugs and medical devices to the U.S.: self-certification by manufacturers for medical devices considered to carry little risk, certification by public or private third parties for higher-risk medical devices and independent review committees for drug approval.
While this would be an improvement, we can do better. We can make drug and medical device certification more responsive, both to researchers trying to introduce new lifesaving drugs and to consumers who need them, by abandoning the FDA system and handing drug certification over to private companies. In a free market for drug certification, competition and legal liability would help to ensure that safe drugs are available faster. The FDA would merely set standards and police the market or would be eliminated.
While many Americans favor privatization of other government functions, some are reluctant to privatize health and safety regulation. How do we know the marketplace will work? Because we know it works to ensure the safety and effectiveness of thousands of other products. For more than a century, Underwriters Laboratories, Inc. (UL) and its competitors have protected American consumers from unsafe and ineffective products. The example of UL shows how private agencies might go about certifying drugs and medical devices.
[page]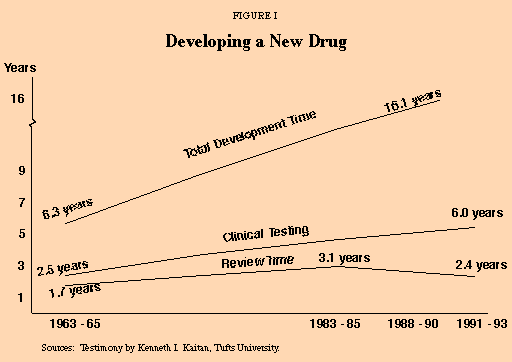
Developing a new drug is a complicated process. After the compound is produced in a laboratory, it undergoes preclinical testing on animals, clinical testing on humans and FDA review. The time required for the process has increased significantly since 1963 [see Figure I].2
- The time required for clinical testing has increased by 140 percent, from 2.5 years in 1963-65 to six years in 1991-93.
- The review time has increased 41 percent, from 1.7 years to 2.4 years.3
- Including preclinical animal testing, the total time from laboratory production of a drug to FDA approval has increased 156 percent, from 6.3 years in 1963-65 to 16.1 years in 1988-90.
"Total time from laboratory production of a drug to FDA approval has increased…to 16.1 years."
How the FDA Delays Drugs. Many of the medical products developed today require longer and more complicated testing because of their complex nature. In addition, despite a well-orchestrated public relations campaign suggesting just the opposite, the FDA imposes delays at every stage of development.
In an October 1995 report, "FDA Drug Approval," the General Accounting Office announced that review times have fallen from 33 months to 19 months.4 In a December 12, 1995 speech at the annual meeting of the Food and Drug Law Institute, FDA Commissioner David Kessler boasted that the FDA had that year reviewed 96 percent of all pending final applications.5 Those numbers are misleading. The GAO report looked only at the last review of a final drug application and ignored the other phases of FDA review. The other phases have lengthened overall development time. For example, the number of clinical trial subjects has increased and the number of tests per subject has doubled. The FDA also has greatly increased the amount of information it demands before accepting a new drug application.6
Jeffrey Kimball, executive director of the Medical Device Manufacturers Association, says the FDA reduced its backlog of pending applications by a simple procedure: it rejected more new drug applications.7 A report by Citizens for a Sound Economy shows the FDA is taking more time to approve fewer drugs. In fiscal year 1994 the average review time was 27.3 months for the 62 new drug applications approved. But in 1995 the 33 approved new drug applications took an average of 28.6 months.8 U.S. law requires review of applications for final approval of new drugs within 180 days.9 The FDA is nowhere near meeting the statutory limit.
What is the result? Extremely long delays between the laboratory development of a drug and the time it is available to patients.10
- According to Sen. Nancy Kassebaum (R-KS), former chairman of the Senate Labor and Human Resources Committee, which has jurisdiction over the FDA, it takes 12 years for drug companies to develop a new drug and gain FDA approval to market it.11
- Kenneth I. Kaitan, associate director of the Tufts Center for the Study of Drug Development, estimates that the total time to develop and gain approval for a new drug is actually 16.1 years.12
The Perils of Delaying Drugs. Even with the best motives and the utmost enthusiasm, the staffs of regulatory agencies make mistakes and cause delays. Often, such mistakes and delays lower businesses' profits, destroy jobs and raise the prices consumers pay. But when the FDA makes mistakes, people die. And when it fails to act or acts too slowly for fear of making a mistake, people also die.
"Some lifesaving drugs were available in other countries years before the FDA approved them for use here."
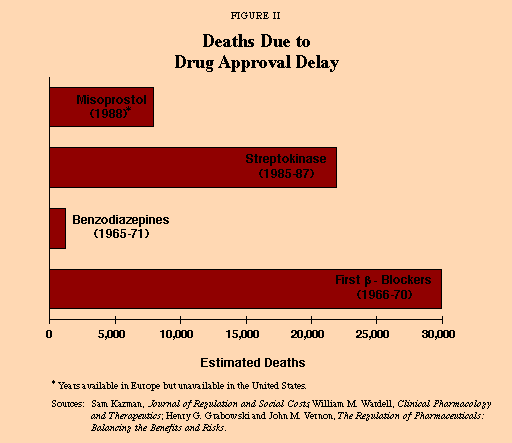
These deaths are not dramatic, and one cannot always identify a death that occurred because the FDA was slow to allow a drug on the market. There is very little peer-reviewed research on deaths caused by FDA delays.13 Nonetheless, to a certain extent quantification is possible based on FDA claims regarding the lifesaving benefits of drugs it approved and the fact that some lifesaving drugs were available in other countries years before the FDA approved them for use here. Some examples [see Figure II]:
- Misoprostol is 94 percent effective in preventing bleeding ulcers, common in arthritis sufferers taking aspirin and similar drugs. According to the FDA, it can potentially help 10,000 to 20,000 people every year. For the nine and one-half months it took the FDA to approve the application for this new drug, no one in the U.S. could use it. That means between 8,000 to 15,000 Americans may have died because Misoprostol was not available sooner.14
- Thrombolytic therapy dissolves blood clots in heart attack victims. Every year 700,000 people suffer heart attacks, and 9 percent of them die. The FDA found the therapy reduced heart attack fatalities by 18 percent, but it took two years to approve the new drug application. The result was up to 22,000 deaths.15
- More than 1,230 U.S. deaths from overdoses of sedatives might have been prevented during the five years a safer hypnotic (benzodiazepine) was approved in Britain but not by the FDA.16
- Beta-blockers effective in preventing heart attacks and coronary death might have saved 10,000 to 20,000 lives a year during the three years when none were available in the U.S.17
- An estimated 1,000 lives were lost when FDA red tape prohibited the manufacturer from selling a defibrillator that it had already approved, according to Dr. Richard Cummins of the American Heart Association.18
These numbers reflect only the fatalities that can be laid at the FDA's door. They say nothing about the pain and suffering. Our quality of life declines when drugs or devices cannot be sold until exhaustive, byzantine FDA approval procedures are complete.19
Approvals Still Delayed. For more than three years the FDA has worked to reduce the amount of time it requires to approve new drugs. The FDA and Vice President Al Gore's National Performance Review proudly predict that by 1997 new drugs will receive final approval and be marketed within one year.20 Ignoring all the delays earlier in the process, what are the consequences of delaying new drugs for only one year? In the case of thrombolytic therapy, that means only 11,000 people might have died.
The FDA also imposes heavy financial burdens on Americans. Some medical manufacturers are responding to the FDA's costly delays by moving abroad. The 16-year time frame for the development and approval process has raised the average cost of drug development in the U.S. to an estimated $200 million.21 Not surprisingly, more and more medical companies are relocating portions of their business, especially to Europe, where a more rational regulatory environment makes drug development less costly.
- In a June 1994 survey by the American Electronics Association of 58 companies producing medical devices, 29 percent of the polled firms indicated they had shifted investment overseas while 22 percent said they had shipped personnel overseas and 40 percent said they had reduced their U.S. payroll as a result of FDA delays.22
- A 1994 survey by the Minneapolis Star Tribune found that the most heavily regulated companies employed 7 percent of their personnel overseas in 1989 and 12 percent in 1994, and expected overseas employment to reach 16 percent by 1999.23
Taxpayers also bear a substantial burden because the FDA is funded mostly by tax dollars, not drug company fees. In 1994 the total FDA budget was $921 million, appropriations for 1995 were $975 million24 and the 1996 budget was $878 million.25
[page]To reduce drug development and approval times, various administrative and legislative reforms have been enacted or proposed.
User Fee Act. The first major reform bill enacted, the 1992 Prescription Drug User Fee Act, allows the use of unapproved drugs for such serious conditions as AIDS and cancer before clinical trials are complete. For other drugs, the act spells out user fees manufacturers can pay to provide resources that speed up application reviews. It also mandates that the FDA meet goals for improving its performance. It is required to review progressively higher percentages of new drug applications within one year – which is still longer than the statutory six-month limit for reviewing applications.
FDA Initiatives. Before his resignation in November 1996, even FDA Commissioner Kessler saw the need for change or at least the congressional threat to the FDA. Kessler had stated, "[W]e are working hard to make the FDA more efficient," and "[W]hen it comes to getting needed therapies to dying patients, the riskiest thing we can do is be unwilling to take risk."26 Thus in 1994 the FDA exempted from approval requirements several hundred low-risk medical devices that are substantially equivalent to devices already on the market.27 By January 1996 the FDA had exempted a total of 572, nearly three-quarters of the lowest-risk devices.28
In August 1996 the FDA initiated a pilot study of a European-style review system for low- to moderate-risk devices. For the study, the FDA has accredited outside reviewers to test the design, performance and safety of at least 10 categories of devices. Unlike the European system, where only certification by an independent reviewer is necessary before a device is marketed, the U.S. pilot study requires the outside reviewer to make a recommendation to the FDA, which makes the final decision.29
"The FDA has accredited outside reviewers to test the design, performance and safety of at least 10 categories of devices."
Senate Proposal. Sen. Nancy Kassebaum's FDA reform bill S.1477, The FDA Performance and Accountability Act, passed the Senate Committee on Labor and Human Resources on a bipartisan vote of 12 to 4.30 It contained common-sense reforms reining in FDA power and reducing the regulatory burden on manufacturers without substantially altering FDA methods. A revised bill with fewer reforms was reported out of the committee on June 20, 1996. The 104th Congress took no further action but the new Congress may consider similar legislation.
S.1477 would have required the FDA to establish an in-agency appeals system with scientific review groups to examine disputes. It would have required the FDA to allow the use of one clinical investigation, instead of two, to demonstrate the safety and efficacy of a drug.31 It also would have required the FDA to make experimental therapies more readily available to patients. The Senate bill also included provisions for these more fundamental changes at the FDA:32
- Performance Standards. The FDA would be required to develop and publish quantifiable performance standards for its own review process.
- Product Performance Standards and Reasonable Data Requirements. The FDA would be required to recognize appropriate device performance standards set by certified, independent organizations. The FDA also would be required to establish criteria for data that must be included in applications.
- Scientific Review Groups. The FDA would have to expand its existing expert review system. The advisory committees would have more independence, and the FDA would respond to their recommendations in a timely manner.
- Third-Party Review. The FDA would have to establish requirements for a third party to become an outside reviewer. Manufacturers could seek third-party review and recommendation for approval of medical devices. The FDA would then have to accept or reject the third party's recommendation.
House Proposals. During the 104th Congress, three FDA reform bills were introduced in the House and referred to the House Commerce Committee.33 Two of the bills called for many of the same reforms as the Senate bill but removed more of the FDA's discretionary power.34 Unlike the Senate measure, the House bills obligated the FDA to harmonize its regulatory requirements with those of other countries.35
The major difference between the Senate bill and the House bills was the role of third-party certifiers. In the House bills, manufacturers could take their drugs or devices to the independent certifier without first seeking FDA approval. The drugs and devices would be assumed approved if the FDA did not respond to the third-party recommendation within 60 days. If the FDA vetoed a recommended approval, it would have to explain why.
[page]"Proposed reforms represent an enormous improvement over the status quo."
Most of the proposed reforms represent an enormous improvement over the status quo. However, they either create problems of their own or do not adequately address FDA failings. Also, bills change significantly as they move through the Congress from committee to floor to conference committee. Often, to win passage, fundamental reforms are deleted. No matter how good a proposal looks, the probability that it would pass Congress and become law in its original form is low.
Increasing the Use of Independent Review Boards. Greater reliance on independent councils comprising researchers and possibly industry personnel to review and approve drug applications would likely have one of two effects. At one extreme, reliance on independent review boards might give industry too much influence over the drug approval process. Researchers and manufacturers deserve a place at the table, but if the FDA is forced to accept the advice of industry-staffed review boards, its independence is sacrificed.
At the other extreme, the FDA might ignore researchers and manufacturers even when they sit on the independent review councils. But if the FDA is not required to respond to the industry, there is no reform at all. The FDA would still be able to engage in the delaying behavior Sen. Barbara Mikulski (D-MD) spoke of when she said, "There is enormous frustration about … enforcement over approval, about nit-picking over a label as compared to really focusing on moving to the marketplace."36
Then there is the middle road for the FDA, the road already taken. This balanced path between industry and FDA monopoly is full of difficulties as well. The researchers and medical professionals who staff the FDA's review boards have little incentive to devote the necessary time and energy. They have other full-time occupations, yet they must know about medical therapies as well as clinical testing theories and practices. And they are supposed to have no conflicts of interest.
The reality is that review panel members may have little if any knowledge of the science behind a new product. And the FDA can and does exercise waivers for panel members who have financial or other conflicts of interest.37
Independent review boards have their own built-in delays. Review boards are required to operate separately from any manufacturer. No cooperation between them during design and production of a product is permitted. This forces the manufacturer to complete all of its work before submitting it to the body that must then review all of the data.
"Congressional proposals would still allow the FDA to block or delay the marketing of drugs."
Increasing the Use of Third-Party Certification. Drug companies could bypass the FDA if they could contract with a third party to certify their test results, then market their drugs. Increasing the number of therapies that can be independently certified could save American lives. This reform would move the U.S. closer to Europe's system of private, often competing, certification bodies. However, the congressional proposals would still allow the FDA to block or delay the marketing of drugs.
And the third-party certification system has the same sort of delay built into it as does the independent review board system. The manufacturer must incur all the time and expense of development. Then it must seek – and perhaps fail to receive – the recommendation of the third party.
Another key defect in the House and Senate plans is the proposed accreditation process for the third-party reviewers. The Senate plan leaves the process up to the FDA commissioner. The House plans spells out accreditation requirements. Authority to set the requirements is a powerful tool, determining the nature, characteristics and behavior of the third-party reviewers. The House plans would invite outside influence, since adjusting or adding to the requirements would be a congressional and therefore a political decision.
Using Performance Standards. Currently, the FDA has no definite standards of performance for safety or effectiveness that a drug can meet to ensure approval. Written standards, harmonized with those of the rest of the world, would be an improvement over current policy. However, government-mandated or approved standards would have a subtle yet profound influence on drug company research. Research would focus on creating drugs that would meet the standards, not simply save lives and ease pain.
Further, FDA-imposed standards, set by the commissioner in consultation with interested parties, would reflect the concerns of the special interests. Protecting the standards from this pressure is too much to ask of any provision limiting conflicts of interest. Harmonizing U.S. standards to international standards might do no more than allow foreign special interests to influence U.S. drug approval procedures.
Finally, FDA-imposed standards would inevitably become politicized. While its proponents might argue that the agency is insulated from special interests, the FDA gets its budget and mandate from Congress, which resists few political pressures.
[page]"The drug approval process could be made faster and more cost-effective without sacrificing the safety and effectiveness of drugs."
In order to determine if proposed FDA reforms will accomplish their purpose, we must consider the general nature of regulatory reform as well as specific proposals. Generally, what Congress deregulates it can reregulate. And it has a history of forcing the political pressure it feels onto the backs of the agencies under it. Consequently, federal agencies are marked by "mission creep." What begins as a simple and understandable mandate for safety and effectiveness becomes a welter of confusing and contradictory congressional demands. Joel Nobel, president of ECRI, an independent medical-device testing company, explains Congress' approach to the FDA:
First require that the FDA do the unwise or impossible. A few years later, ask the General Accounting Office to tell you if the FDA is doing the unwise or impossible as instructed. Express shock and surprise when you learn that it is not. Hold hearings to pistol-whip the FDA and industry in order to support the passage of more unwise or impossible-to-implement legislation.38
The drug approval process could be made faster and more cost-effective without sacrificing the safety and effectiveness of drugs. How? By moving to a market-based system in which no one could block drugs from entering the market, and no one could prevent consumers from making their own decisions about the drugs they used. The FDA would have no substantial role, as private third parties would certify drugs and devices. This system is not only possible, but preferable.39
Privatizing government functions is now commonplace. Yet questions and fears remain. We can address them because in this instance we know what is likely to happen when the FDA no longer compels premarket approval. Our guide is an institution that has certified safety for more than 100 years: Underwriters Laboratories, Inc., an independent, not-for-profit safety certifications and standards-writing organization.
[page]Like the FDA, Underwriters Laboratories, Inc. is committed to public safety. Both organizations work to safeguard the public from dangerous products. Both are staffed by experts in science and technology. UL's actions affect millions of consumers and billions of dollars, as do the FDA's. Every day, U.S. consumers buy and use products that are "UL Listed," meaning UL certifies that they meet its standards, or FDA-approved, meaning that they meet FDA standards.
"Unlike the FDA, Underwriters Laboratories cannot deny consumers choice."
But there are differences. UL listing means that experts have given their opinion on the safety of a product. FDA approval is a legal requirement before drug and medical device companies can market their products. Unlike the FDA, UL operates a wholly voluntary certification system and cannot compel manufacturers to submit products for testing. Unlike the FDA, UL cannot prevent products from being marketed. It cannot deny consumers choice because it is not a regulatory agency with enforcement powers. Unlike the FDA, UL receives no tax revenues. It is wholly funded by its clients, mostly manufacturers, but is independent from them.
Yet every day we put on our FDA-approved cosmetics after drying our hair with our UL Listed hairdryers. We cook our FDA-approved breakfast cereal on our UL Listed stove top. On our drive home, we take our FDA-approved aspirin while cooling off with our UL Listed automotive air-conditioner. At night, we sleep soundly, protected by our UL Listed smoke detector. Both UL and the FDA contribute to our safety and peace of mind.
What UL Does. Since 1894, UL's stated mission has been "Testing in the Public Interest." William Henry Merrill, its founder, said in 1923, "We are doing something for manufacturers, buyers, users, and property owners everywhere. We are doing something for humanity."40 [See the Sidebar on Underwriters Laboratories.]
UL conducts product safety and performance testing and certification as well as quality system registration. It also develops national and international codes and standards. Along the way, UL writes Standards for Safety that manufacturers can use to aid them in designing and marketing safe, salable products.
The only business of Underwriters Laboratories is providing information to the public. UL underwrites no risk in the insurance industry sense. It produces no testimonials, advertisements or other marketing support. UL approval requires manufacturers to issue warning labels, use-and-care booklets, safety tips and other consumer information. UL itself distributes informational literature, broadcasts public service announcements and publishes news releases to educate the public about the meaning of the UL mark.
No Delays, No Politics, No Coercion, No Monopoly. UL cannot be accused of delaying the approval of new products because it cannot keep products off the market. UL tests products and certifies their safety, providing consumers with accurate, timely information and nothing more. Consumers can decide for themselves, based on good information, whether they want to buy a riskier product or not.
Most of us gain from choices other people make on our behalf using UL's services. Based on UL approval, builders install household chimneys and fire-resistant wire for our benefit. No monopolistic government agency prevents us from making choices or deters others from making good choices on our behalf. The UL system has worked for more than 100 years!
UL demonstrates a way to assure third-party independence. Its standards are not created in a political furnace. They do not reflect political or industry special interests.
"The UL System has worked for more than 100 years."
No statutory, regulatory or court-ordered mandate requires manufacturers to seek UL approval, yet tens of thousands do. Why? Because buyers want safe and effective products. UL has staked its reputation, time and continued existence on the accuracy and timelines of the information it provides. Consumers recognize this and are willing to buy, or pay more for, UL Listed products. Thus manufacturers who produce a good product want UL listing. Companies that make poor, unsafe products are not listed with UL, and many consumers avoid these goods. To know a product is safe, all the consumer has to do is look for the UL mark.
Manufacturers pay for UL services. UL charges a fee based on the cost of testing and bills for its on-site inspectors at a flat rate. Consumers who do not benefit from its services do not have to pay for them. Since it does not receive a government appropriation, UL is not pressured to comply with congressional special interests. On the other hand, UL must satisfy customers directly and is not detached from the people who use its services as are tax-supported government agencies.
If UL were merely a tool of certain manufacturers, it could not avoid listing unsafe products. Consumers would soon discover this, and the UL mark would be worthless. Manufacturers would stop paying for UL and its services. UL, consumers and manufacturers all want the organization to remain accurate and independent, and all have incentives to guarantee it.
UL does its job efficiently and at low cost. In 1994, UL employed more than 3,900 people, including more than 900 degreed engineers and many researchers and technicians. In that same year, the FDA employed 1,093 people in Commissioner Kessler's office alone.41 Revenue figures were not publicly available, but in 1994 UL had total current assets of $61,714,000.42 That is a substantial sum, but far less than appropriations for the FDA.
UL takes an active, long-term role in quality and safety assurance. For their fees, clients receive many follow-up services. These include frequent, unannounced visits to production facilities worldwide. In 1994, UL personnel conducted more than 481,000 on-site, follow-up service visits.43 During a visit, UL personnel check production controls, observe on-site testing, inspect facilities and select samples for further testing at UL labs. They even check to see if the Certification Program is posted on the wall. If the facility does not pass inspection, the manufacturer has two weeks to correct the mistakes. After that, UL pulls its certification.
Even Governments Use UL. UL also has federal agencies, states and localities as clients. For example:
- The Occupational Safety and Health Administration uses UL as a tester and independent certifier of "hazardous location" products, and the organizations work together to create, revise and implement standards for products falling under OSHA's authority. In some cases, OSHA uses UL's existing standards.44
- UL works with more than 40,000 jurisdictions across the U.S. to develop electrical, building and safety codes, and the UL mark is accepted in all 40,000.45
How a Standard for Safety Is Written. UL is often involved with product researchers and developers from the beginning. Shortly after the initial design work is done, manufacturers bring UL in so that standards are written and a Certification Program is in place by the time the good is actually produced.
"Close cooperation between the producers and UL allows faster production and certification of safe products."
This close cooperation between the producers and UL allows faster production and certification of safe products. For example, Scott Ritchie, Tampa Local Engineering Service Manager, conducted safety and performance tests during a tropical storm. Gordon Beamer of ECC International of Orlando, Fla., said "I just can't believe he made it here during Tropical Storm Gordon….I wanted to make sure the new design was acceptable as quickly as possible. The people here couldn't believe Scott even got here. We were closing the plant and he was running tests."46
After a product has been submitted for testing, UL issues an outline of investigation to interested parties and solicits comments and criticism. Based on the feedback, UL amends the outline, issues the proposed standard and repeats the process. This reiteration results in the published standard. The whole process usually takes three to four months. Amendments to the standard can be published and available within a day, and UL's engineers can write a Certification Program in two weeks.47
The standards are designed to accommodate manufacturing innovation in a manner consistent with the standard's original intent. The guide to developing standards says, "[B]uilt into UL Standards are requirements that facilitate changes and eliminate undue restrictions on design. This equivalency makes it possible to convert any construction requirement into a performance requirement."48
Comparing the UL Approach and the FDA Approach. The FDA does not have published standards, nor does it tell manufacturers how to comply with requirements to get a new drug approved.49 Instead, it tells the manufacturer to hire a consultant to guide it through the process.50
FDA Commissioner Kessler sought to increase FDA flexibility by allowing manufacturers to slightly alter the content or manufacturing process of a drug or biologic with only a brief notice to the agency instead of a full review.51 Also, the FDA plans to implement a policy that certifies safe manufacturing processes rather than specific product performance.52 Responding to the increased pace of technological innovation, UL already helps companies register facilities and quality systems worldwide.53 In other words, while the FDA is only beginning to concern itself with quality production systems, UL has already done so.
"While the FDA is only beginning to concern itself with quality production systems, UL has already done so."
The Assurance of Good Work. During testimony before the Senate Committee on Labor and Human Resources, Kessler stated, "The assurance that the FDA is there every day, doing its job, is so fundamental that we have the luxury of taking it for granted." This statement implies that the FDA is necessary for Americans to feel secure about their food and drugs. However, Americans do not worry that their televisions will start fires or that they will be injured using their toasters. The reason is that UL is conscientious and accurate and has incentives to stay that way.
Poor quality and poor performance standards are costly for everyone, including manufacturers and UL. As UL's 1994 Annual Report states, "The 'real' cost…is compromised safety, which can ultimately result in product rejection, manufacturing delay and greater costs. A final result is the loss of the certification organization's credibility and the manufacturer's product acceptance." That means loss of clients for UL and loss of jobs for UL's management and employees – good reasons to maintain high standards.
Furthermore, UL does have competitors. They include Electronics Testing Laboratories of Cortland, N.Y., a subsidiary of the British conglomerate Inchcape, Factory Mutual of Norwood, Mass., and Canadian Standards Association of Rockville, Ontario. Some use UL standards as the basis of their certification, but others write their own. Though UL uses the term "friendly competition,"54 the competition is there. If UL's standards are inappropriate or if the consuming public loses confidence in UL, others are ready to serve the market.
[page]Because of the work of UL and its competitors, we believe that bulletproof glass is indeed bulletproof and that our smoke detectors will indeed wake us in the event of a fire. But Zantac, amoxicyllin and cardiac arrest paddles are very different from light bulbs, toasters and cordless telephones. Can we expect a certifying organization modeled after UL to protect us when drugs and medical devices are involved? There are four concerns about private certification that we must address.
Protecting Consumers from Dangerous Drugs. To get straight to the heart of the major consumer concern, can a company modeled after UL protect us from thalidomide? Contrary to popular belief, the FDA was not responsible for preventing thalidomide tragedies in the United States, nor was the drug approval process in Europe riskier than in the U.S.55 We can be protected to the extent that we want to be protected. A third-party certifier has no reason to list a drug unless it is sure of the safety of the drug. Under such a system, a dubious drug might be on the market, but it would lack third-party certification. Doctors, mindful of both the Hippocratic Oath and the state of medical liability, would be reluctant to prescribe medicines that lacked such a listing. Only consumers willing to carry a great deal of risk would use such a drug – and they likely would do so over their doctor's objections.
Private certification would allow different people in different circumstances to balance the risks they accepted. In concrete terms, no bureaucrat could prevent someone dying of AIDS from buying and using AZT or new experimental drugs.
"No bureaucrat could prevent someone dying of AIDS from buying and using AZT or new experimental drugs."
Providing Consumers with Effective Drugs. UL and other companies conduct performance tests only for life-safety products such as smoke alarms and fire extinguishers, but the FDA must meet the performance mandate on every application. Can a private company accommodate performance testing on a much larger basis? Yes. The certification industry would change but the certifying organizations could adapt. Adding a performance mandate would not alter the market incentives for third-party organizations. They probably would still be involved in research and development from the earliest stages and probably would still produce flexible, adaptable standards. Importantly, the third-party companies would still deal directly with their final clients: consumers and manufacturers.
Preventing a Race to the Bottom. Competition in the private market creates powerful incentives to reduce costs. Would private competition in the market for drug certification result in a reduction in the quality of work as a way to keep costs down? The history of UL and its competitors in the certification business assures us that it would not. As competing organizations have come into the market, the testing burden has not become easier for manufacturers, and consumers remain confident in the safety of their products. What has happened instead is that new market entrants have adopted the efficient, accepted UL standards and competed on cost or personal service.
Manufacturers must now have the FDA's permission to test new drugs on humans after completing the required animal studies. However, such tests may not uncover the risks of long-term, widespread use.56 Under competitive drug certification, the efficacy and extent of human testing and the best way to reduce the health risks to test subjects would be scientific questions on which certifiers, manufacturers and their medical advisers could reach a consensus. Today, whether patients are allowed to receive unapproved drugs during the testing phase is a political decision; under a market-based system, patients and other test subjects could receive uncertified drugs if they were willing to assume the risk.
Providing a Stable Certification Program. Expanding third-party certification into drugs and medical devices – matters of life and death – would increase the liability of the certifying organizations. But they could adapt to a greater level of risk. Third-party certification simply states the professional opinion of the certifiers as to the safety and, perhaps, effectiveness of the product. Certifiers can be sued, but judicial interpretations of liability do not assign liability to certification organizations like the ones under discussion. Third-party medical certifiers would not suffer devastating judgments against their assets or be paralyzed by concern over their exposure. The law would still protect consumers. While certifiers would be protected from liability, injured consumers could seek legal redress from manufacturers and distributors.
[page]"Congress should abolish the FDA and allow private organizations to certify drugs and medical devices."
The FDA badly needs reform. Consumers know it; manufacturers know it; Congress knows it; even the FDA knows it. Almost any reform would represent an improvement over the status quo. How do we best alter a vast system that has existed for decades? A number of thoughtful and laudable proposals have been discussed in Congress, most of them centered on making the FDA efficient and accountable. However, the proposals all would preserve the FDA's insulation from consumers and manufacturers. The FDA would remain subject to congressional pressure. It would retain the power to prevent new drugs from entering the market or could regain the power to do so.
Why not have real reform and turn the entire process over for private, third-party certification? We would get safe and effective drugs, quickly and efficiently. We would be free to choose our own level of risk. We would be able to relieve pain without fear for our safety.
Thus the best reform is the most comprehensive reform. When the 105th Congress undertakes FDA reform, the reform should be profound. Congress should abolish the FDA and allow private organizations to certify drugs and medical devices.
NOTE: Nothing written here should be construed as necessarily reflecting the views of the National Center for Policy Analysis or as an attempt to aid or hinder the passage of any bill before Congress.
[page]- The author thanks the people of Underwriters Laboratories, Inc. for their unstinting assistance far beyond the call of common courtesy. The author thanks especially Frank Brutomesso, Judy Lykins, Homer Pringle and Rick Titus. All errors, inferences and comparisons to the FDA are solely the responsibility of the author and in no way reflect the attitudes or opinions of Underwriters Laboratories, Inc.
- Kenneth I. Kaitan, associate director, Tufts Center for the Study of Drug Development, Tufts University, written testimony for the U.S. House Subcommittee on Oversight and Investigations, May 25, 1995.
- The 2.4 years in 1991-93 was down from a high of 3.1 years in 1983-1985.
- United States General Accounting Office, "FDA Drug Approval," October 1995.
- David Masci with Steve Langdon, "Bill to Overhaul FDA Process Gets Nod from Panel," Congressional Quarterly, March 30, 1996, p. 887.
- Robert M. Goldberg, adjunct scholar, American Enterprise Institute, "Why Kessler Must Go," Wall Street Journal, April 4, 1996.
- Steve Langdon, "FDA Drug Approval Process May Undergo Surgery," Congressional Quarterly, January 27, 1996, p. 223.
- Lydia Verheggen, "FDA Review Times: Not Making the Grade," Issue Analysis No. 23, Citizens for a Sound Economy, February 26, 1996, p. 3.
- 21 USC 355(c)(1).
- Sam Peltzman "conservatively" estimated that the 1962 amendments to the Food, Drug and Cosmetic Act, which required that new drugs be proven effective – not just safe – added a minimum of two years to the drug approval process. See Peltzman, Regulation of Pharmaceutical Innovation: The 1962 Amendments (Washington: American Enterprise Institute for Public Policy Research, 1974), p. 18.
- Sen. Kassebaum is quoted in Masci and Langdon, "Bill to Overhaul FDA Process Gets Nod from Panel." The Biotechnology Industry Association makes a similar estimate; see Pete Du Pont, "National Nanny on the Ballot," Washington Times, July 17, 1995.
- Testimony of Kenneth I. Kaitan.
- Conversation with Robert M. Goldberg, senior research fellow at George Washington University Medical School's Center on Neuroscience and Medical Progress, November 12, 1996.
- Sam Kazman, "Deadly Overcaution: FDA's Drug Approval Process," Journal of Regulation and Social Costs, August 1990, p. 43. The fatality figures were calculated by multiplying the length of time required to approve the new drug application by the number of lives the FDA claimed the therapy might save.
- Kazman, "Deadly Overcaution," p. 44.
- William M. Wardell and Louis Lasagna, Regulation and Drug Development (Washington: American Enterprise Institute, 1975), ch. VII, p. 73; an updated and condensed version of W. M. Wardell, "Introduction of New Therapeutic Drugs in the United States and Great Britain: An International Comparison," Clinical Pharmacology and Therapeutics, vol. 14, 1973, pp. 773-90.
- Wardell and Lasagna, Regulation and Drug Development, pp. 111-12; and U.S. Congress, House Subcommittee on Science, Research and Technology of the Committee on Science and Technology, "Report on the Food and Drug Administration's Process for Approving New Drugs," 96th Congress, 1st session, 1980, p. 32, cited in Henry G. Grabowski and John M. Vernon, The Regulation of Pharmaceuticals: Balancing the Benefits and Risks (Washington: American Enterprise Institute, 1983), p. 46.
- Interview with Dr. Richard Cummins, ABC News, "20/20," August 12, 1994, cited by Alan M. Slobodin, "The Real Problems with Health Care in America: While Dr. David Kessler's FDA Fiddles, Medical Approvals Lag and Americans Die," Legal Backgrounder, vol. 9, no. 36, October 28, 1994, Washington Legal Foundation, p. 2.
- Telephone conversation with Dr. Robert Higgs, director of the Independent Institute, June 14, 1995. Higgs emphasizes the convoluted nature of U.S. drug approval.
- President Bill Clinton and Vice President Al Gore, "Reinventing Drug and Medical Device Regulations," National Performance Review, April 1995, pp. 2-3.
- Carolyn Lockhead, "Deadly Over-Caution: FDA Assailed for Slow Testing of New Drugs;" San Francisco Chronicle, October 26, 1992, quoted in Michael I. Krauss, "Loosening the FDA's Drug Certification Monopoly: Implications for Tort Law and Consumer Welfare," George Mason Law Review, Spring 1996, p. 463.
- American Electronics Association News Release, Washington, DC, June 23, 1994, quoted in Robert Higgs, "Wrecking Ball: FDA Regulation of Medical Devices," Cato Institute Policy Analysis No. 235, August 7, 1995, p. 34.
- Ibid., p. 34.
- Budget figures are from "Fiscal Year 1996 Justification of Estimates for Appropriations Committees for Food and Drug Administration," February 6, 1995, p. 9.
- Steve Langdon, "FDA Drug Approval Process May Undergo Surgery," Congressional Quarterly, January 27, 1996, p. 222.
- Ibid., pp. 11, 24.
- These 510(k) approval requirements are different from the premarket approval for a new device or new use, which requires the full approval process.
- Telephone conversation with Paul Tilton, Center for Devices and Radiological Health, FDA, April 26, 1996.
- Clinton and Gore, "Reinventing Drug and Medical Device Regulations," p. 17, and telephone conversation with Al Bracey, Division for Small Manufacturers' Assistance, Center for Devices and Radiological Health, FDA, September 18, 1996.
- Press release from Sen. Kassebaum's office, "Labor Committee Passes Comprehensive Reform of FDA with Strong Bipartisan Support," March 28, 1996.
- Proceedings, Senate Committee on Labor and Human Relations, April 6, 1995, Hearings On the Line, pp. 2-8.
- "A Summary of S.1477 as passed by the Senate Committee on Labor and Human Resources on March 28, 1996," Senate Labor and Human Resources Committee, p.1, pp. 2-8
- Telephone conversation with Anthony Habib, staff member for the House Commerce Committee, April 26, 1996. The bills are: Rep. Richard Burr (R-NC), H.R.3199, The Drug and Biological Products Reform Act; Rep. Joe Barton (R-TX), H.R.3201, The Medical Device Reform Act of 1996; and Rep. Scott Kluge (R-WI), H.R.3200, The Food Amendments and Animal Drug Amendments of 1996.
- All information is from the House Commerce Committee's section-by-section analyses of The Medical Device Reform Act of 1996, and The Drug and Biological Products Reform Act, provided to the author by Anthony Habib, April 26, 1996.
- "The Medical Device Reform Act of 1996: Section-by-Section Analysis," House Commerce Committee, p. 1.
- Proceedings, Senate Committee on Labor and Human Resources, April 6, 1995.
- Open letter to Congressman Wayne T. Gilchrest from Jeffrey N. South, Washington Times, March 13, 1996.
- Alexander Volokh, "Clinical Trials – Beating the FDA in Court," Reason, May 1995, p. 24.
- For drugs that aren't otherwise controlled because they are illegal, addictive or abused, the FDA determines whether a patient must have a prescription from a physician to purchase or use them. Under the system of private certification, patients would be free to purchase certified or uncertified drugs without a doctor's advice or permission and assume the risk – although manufacturers might want to restrict sales to physicians or to customers with prescriptions, and they would of course be liable for their own negligence. A comparison of the United States to countries with less regulated access to medicines concluded that the less regulated markets are no less safe – suggesting that regulation of drug use by prescription is ineffective, or no less effective than cautious consumers acting on their doctor's advice. See Sam Peltzman, "The Health Effects of Mandatory Prescriptions," Journal of Law & Economics, October 1987, pp. 207-38.
- W. H. Merrill is quoted in This Inventive Century: The Incredible Journey of Underwriters Laboratories (Northbrook, IL: Underwriters Laboratories, Inc., 1994), p. 5.
- Senate Committee on Labor and Human Resources, April 6, 1995, Hearings on the Line, p. 61.
- 1994 Annual Report, Underwriters Laboratories, Inc., (Northbrook, IL, Underwriters Laboratories, Inc.), p. 24.
- 1994 Annual Report, Underwriters Laboratories, p. 16.
- Telephone conversation with Al Abadir, OSHA employee, June 15, 1995.
- 1994 Annual Report, Underwriters Laboratories, p. 10.
- Ibid., p. 15.
- Telephone conversation with Frank Brutomesso, Underwriters Laboratories, June 27, 1995.
- "Method of Development, Revision and Implementation of UL Standards for Safety," Underwriters Laboratories, Inc., pp. 10-11.
- See Kazman, "Deadly Overcaution," and Robert Higgs, "How the FDA is Causing a Technological Exodus," Competitive Enterprise Institute, February 1995.
- Telephone conversation with Dr. Robert Higgs, June 14, 1995.
- Proceedings, Senate Committee on Labor and Human Resources, April 6, 1995, p. 10.
- Clinton and Gore, "Reinventing Drug and Medical Device Approval," p. 7.
- "UL – Testing for Public Safety: An Overview of Underwriters Laboratories," Underwriters Laboratories, Inc., p. 6.
- Telephone conversation with Homer Pringle, UL attorney, June 24, 1995.
- Thalidomide, a prescription sedative, was moving toward FDA approval in 1960. Approximately 2.5 million sample tablets were in the hands of U.S. physicians when the manufacturer announced that pregnant women in Europe taking the drug were giving birth to children with deformities. At the time, scientific knowledge of and testing for such side effects was limited, and the FDA had no special expertise in the area. After months of delay, the FDA acted decisively only when President Kennedy insisted. See Robert M. Goldberg, "Breaking Up the FDA's Medical Information Monopoly," Regulation, vol. 18, no. 2, 1995, p. 43.
- Wardell and Lasagna, Regulation and Drug Development, pp. 139-140.
Noel D. Campbell is a doctoral candidate in economics at George Mason University, from which he received a master's degree. He holds a bachelor's degree from Texas A&M University.